MetaProClust-MS1 can cluster metaproteomics using MS1 profiles only
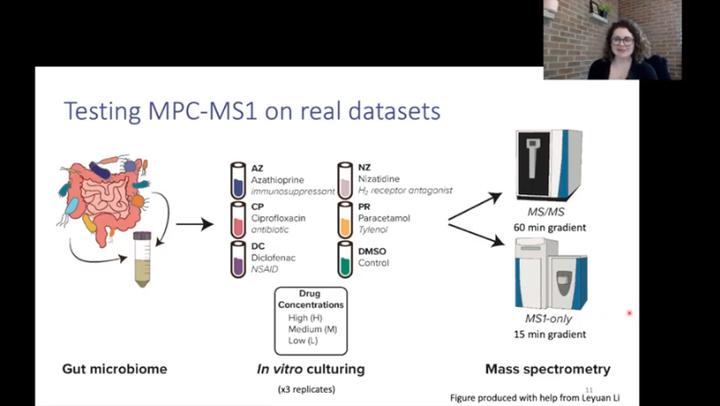 Image credit: YouTube
Image credit: YouTubeAbstract
The human gut microbiota is composed of bacteria, viruses, fungi and archaea that inhabit the digestive tract. These microbes play essential roles in the health and well-being of humans, for example, by fermenting dietary fibres into short chain fatty acids, synthesizing vitamins and contributing to the immune system. Therefore, it’s no surprise that the composition and dysfunction of the gut microbiome is also associated with many diseases, such as inflammatory bowel disease, diabetes, as well as cardiovascular disease and mental health disorders. Recently, studies have shown that human-targeted drugs may significantly disrupt the gut microbiota, and thus there is an increased need to characterize how xenobiotics may be affecting microbiomes and consequently human health. Metaproteomics explores the composition and function of microbial communities and has been previously used to investigate how xenobiotics can affect the human gut microbiome. However, acquiring data by tandem mass spectrometry, the traditional approach to metaproteomics, is time consuming and resource intensive. As metaproteomic experiments become larger-scale, for example as seen in microbiome-drug screens, the resources required for a study also increase. To mediate this challenge, we present MetaProClust-MS1, a computational framework for microbiome screening that reduces the time required for data acquisition by mass spectrometry. MetaProClust-MS1 performs a clustering analysis of samples processed using MS1-only mass spectrometry. In this pipeline, peptide features quantified from 15 minute mass spectrometry gradients are first matched across all mass spectrometry runs from a set of experiments. The approach preprocesses metaproteomics data and performs matrix decomposition using a robust independent component analysis (ICA). K-medoids clustering is then used to further reduce feature dimensionality. Eigenfeature values from peptide intensity values are computed and used as a summary statistic to calculate correlations with microbiome treatments for treatment clustering. The modular, platform-independent code is implemented in R and Python and can be run completely within R Studio. In a proof-of-concept study, we tested MetaProClust-MS1 on a gut microbiome treated with five drugs with known effects at three different concentrations. The samples were analyzed twice: once with short 15 minute MS1-only mass spectrometry gradients and again by a 60 minute tandem mass spectrometry approach. We compared the clusters identified by the framework using both datasets and found that MetaProClust-MS1 identified robust microbiome shifts caused by xenobiotics. A cophenetic correlation coefficient indicated the drug treatment clusters identified using both datasets were also significantly correlated (r = 0.625). Our findings indicate that MetaProClust-MS1 is able to rapidly screen microbiomes using only short MS1 profiles and identifies treatment clusters very similar to those identified using a traditional MS/MS approach. We developed MetaProClust-MS1 for rapid metaproteomic screens to prioritize samples or treatments of interest for deep metaproteomic analysis using more lengthy tandem mass spectrometry experiments. However, MetaProClust-MS1 is not limited to metaproteome screens. For example, the inclusion of feature modules identified by K-medoids clustering also allows for feature module exploration and potential MS1-only biomarker discovery. We anticipate that MetaProClust-MS1 could be extended for protein identification through MS1 features using an MS1-only search workflow. In its currently implementation, MetaProClust-MS1 will be especially useful in large-scale metaproteomic screens or in clinical settings where rapid results are required.
Caitlin Simopoulos
PhD, Sr. Computational Biologist
I use computational biology and machine learning to understand the world around me!